再生障碍性贫血(AA)的诊断和治疗
一、AA的诊断
在决定治疗前,明确诊断是十分重要的。明确诊断后还要①区分AA是获得性还是先天性;②病因;③是否存在异常克隆及异常克隆的数量;④疾病的严重程度。
1、明确诊断:大多数AA的诊断并不困难,但其他疾病也可表现为全血细胞减少和骨髓增生减低,类似于AA,主要是低增生性MDS。制备良好的血涂片和骨髓涂片,取材良好的骨髓活检标本是诊断所必需的。当最初骨髓检查不确定或样本量不够时,为明确诊断需要重复行骨穿检查。


2、除外遗传性骨髓衰竭综合征(IBMFS):所有AA均需要除外迟发型遗传性AA的可能。如果不能正确诊断遗传性骨髓衰竭综合征(IBMFS),会导致①错误地应用免疫抑制治疗(IST);②延误骨髓移植的进行;③错误的BMT预处理方案,导致危及生命的并发症和死亡。正确诊断首先要仔细询问家族史,是否有类似疾病成员和血液系统疾病甚至血细胞数量异常的成员。
30%的Fanconi贫血(FA)患者不伴有身体畸形,临床表现呈多样化,导致在患者成年后才明确诊断。因为Fanconi贫血不能及时确诊,年轻的AA、MDS/AML和上皮细胞肿瘤患者需要除外Fanconi贫血可能,需要进行染色体断裂试验(chromosomebreakagetest/DEBtest)和相关基因的检测。
先天性角化不良(DKC)患者出现典型的临床表现如甲营养不良、网状皮肤色素沉着和粘膜白斑时,诊断并不困难。但5%左右的成人获得性AA存在TERC和TERT基因突变,这部分患者并没有先天性角化不良的临床表现,并对IST治疗无效(隐匿性先天性角化不良)。
少数情况下,Shwachman-Diamond综合征可表现为成人AA的临床表现。身材矮小、干骺端关节畸形、粒细胞减少史、胰腺外分泌功能缺陷,或存在血细胞减少家族史。监测Shwachman-Bodian-Diamond综合征基因(SBDS)突变能发现儿童期漏诊的病例。
3、寻找相关异常克隆:所有患者均应监测异常染色体克隆,至少10%MDS或AML患者在没有形态异常前,存在异常克隆。这部分异常克隆是否代表AA,还是低增生性MDS还存在争论。最近来自英国的研究发现12%的成人AA存在异常染色体克隆。三体(+6、+8、+15)是最常见的异常,通常克隆数量较少。对于三体的患者,对IST治疗的反应、反应的时间和晚期克隆改变的危险性与无染色体克隆改变的AA患者没有明显差异,但非数量核型异常对IST治疗反应差。髓如果存在-7,应该诊断MDS。
流式细胞仪诊断价值已经超过了Ham实验和蔗糖溶血实验,它可以敏感地检测红细胞、中性粒细胞和单核细胞上GPI锚连蛋白克隆数量。许多AA患者能够检测到少量PNH克隆(其百分率受所应用流式细胞仪敏感性的影响),这有助于预测IST的治疗效果。
4、明确疾病严重程度:AA严重性仍采用Camitta(SAA)和Bacigalupo(VSAA)标准。这些简单但实用的标准在决定治疗方案中具有重要价值,并具有一定的预后价值。
再生障碍性贫血(AA)的诊断流程
1.AA诊断标准:
(1)全血细胞减少(至少符合下列两项:①Hb<10g/L;②血小板<100×109/L;③中性粒细胞<1.5×109/L。
(2)骨髓增生减低。
(3)造血组织为脂肪细胞替代。
(4)无异常浸润和网硬蛋白(reticulin)增生。为明确诊断需要进行如下检查:血常规、网织红细胞计数、外周血形态、骨髓穿刺和活检。
2.除外其他疾病导致的全血细胞减少和骨髓衰竭,如低增生性MDS/AML,T-大颗粒细胞白血病(T-LGL),骨髓纤维化(MF)、淋巴瘤、非典型分支杆菌(NTM)感染和神经性厌食症(AN)。
3.除外遗传性骨髓衰竭综合征(IBMFS),如
(1)Fanconi贫血(FA):存在caféaulait点,身材矮小,上肢异常,外周血淋巴细胞加入双环氧丁烷(DEB)/MMC后染色体易断裂;
(2)先天性角化不良(DKC):甲营养不良,粘膜白斑,皮肤色素沉着,肺纤维化,骨质疏松,肝功能异常。DKC-1(X-连锁)、TERC(常染色体显性)、TERT基因突变分析;
(3)Shwachmann-Diamond综合征:胰腺功能不全病史,AA前出现中性粒细胞减少,身材矮小,SBDS基因突变分析。
4.病因学
70%~80%为特发性,没有明确病因。肝炎后AA检测肝功能、病毒检测(包括肝炎A、B、C、G,但通常为阴性,很少数情况下EBV阳性)。药物和化学物质导致的AA,需仔细追问药物和化学物质暴露史,但目前没有明确的试验可以加以验证。阵发性睡眠性血红蛋白尿症(PNH)可检测红细胞、中性粒细胞和单核细胞表面GPI锚连蛋白。
5.是否存在异常克隆
(1)PNH:流式细胞仪检测GPI锚连蛋白。
(2)细胞遗传学克隆:BM细胞遗传学+FISH
(3)T-LGL克隆:TCR基因分析和免疫表型(CD3+、CD8+、CD57+)
6.疾病的严重程度
(1)重型再障(Camitta标准):骨髓增生程度<25%或25%~50%并且造血细胞<30%,并且符合如下三项中至少两项:中性粒细胞<0.5×109/L,血小板<20×109/L,网织红细胞<20×109/L。
(2)极重型再障:符合以上标准,且中性粒细胞<0.2×109/L。
二、获得性AA的治疗
成人再生障碍性贫血(AA)治疗方法主要包括造血干细胞移植和免疫抑制治疗。整体而言,造血干细胞移植和免疫抑制治疗的疗效大致相同。对于20岁以下患者,尤其是VSAA患者,造血干细胞移植的疗效较好。而年龄大于40岁的患者联合免疫抑制治疗的疗效要优于造血干细胞移植。对于20~40之间的患者治疗的选择还存在争论,目前主要根据患者整体情况、疾病的严重程度、患者的意愿和诊治医生对于各治疗方法的熟悉程度。
我国在再障治疗方面有很丰富的经验。对于再生障碍性贫血的治疗,我国杨崇礼教授等在多年工作经验中总结出七项行之有效的治疗原则:
(1)重视支持治疗:主要为贫血和出血的纠正,感染的及时控制。
(2)分型治疗:再障首先要研究类型,慢性再障一般用支持治疗、雄激素和环孢素治疗;而重型再障采用骨髓移植或联合免疫抑制治疗。
(3)早期诊断、早期治疗:大量资料表明病史短者治疗疗效好,病史时间越长,疗效越差。
(4)坚持治疗:治疗方案确定后,至少应该坚持治疗6个月,切忌疗程不足而频繁换药。
(5)维持治疗:国内外大量资料表明,治疗时间短与高复发呈正相关。因此维持治疗应至少在2年以上。
(6)联合用药治疗:无论是慢性再障还是重型再障,联合治疗疗效均优于单药治疗。
(7)合并症治疗:在再障治疗过程中,及时处理各种合并症十分重要。主要为感染的及时控制。

1、联合免疫抑制治疗
抗胸腺细胞球蛋白/抗淋巴细胞球蛋白(ATG/ALG)联合环孢素(CsA)是目前联合免疫抑制治疗的标准方案。它适用于以下患者:①非年轻的SAA患者;②年轻SAA但无HLA匹配同胞供者;③NSAA依赖输血者。IST疗效与病因学关系不大。ATG联合CsA治疗的有效率要高于单纯应用ATG或CsA。具体用法为:马ATG(10~15mg/kg/d×5d),或兔ATG(3~5mg/kg/d×5d)联合CsA(3~6mg/kg/d,至少6个月)。NIH和欧洲多中心研究表明其有效率70%~80%,5年生存率80%~90%。ATG治疗反应一般发生于6个月内,通常在1~2个月可观察到病情的好转,2~3个月脱离血制品输注,但也有较晚起效者。IST有效者应继续服用CsA,逐渐减量至可维持满意血细胞水平的最小剂量,早期或骤然停用CsA可致病情加重或反复。加用粒细胞集落刺激因子(G-CSF)可缩短粒细胞缺乏期。
ATG具有细胞毒性免疫抑制作用,通过清除介导异常免疫的活化细胞毒性T淋巴细胞,解除其对HSC的抑制作用;同时,ATG亦是一种免疫刺激剂,具有致丝裂原作用,能促进淋巴细胞分泌造血细胞生长因子(HGF),如白细胞介素-3(IL-3),粒-巨噬细胞集落刺激因子(GM-CSF)的合成与释放。另外,ATG还能直接作用于HSC表面受体如CD45RO等,刺激其生长或诱使它对HGF的敏感性增高。在低浓度时,能诱导T淋巴细胞表达Fas蛋白和其配体而凋亡;或使T淋巴细胞表面受体与其共刺激分子交联而导致免疫不应答。ATG在体内还可以降低再障骨髓中CD34+细胞Fas抗原的表达。在体外,ATG还可直接促进再障骨髓中CD34+细胞的CFU-GM和BFU-E形成,且所需浓度与ATG使用后体内持续的浓度相当。
环孢菌素A(cyclosporineA,CsA):早先认为CsA与环啡啉(cyclophilin)结合后可抑制钙神经蛋白(calcineurin)对NFAT(nuclearfactorofactivatedTcells,NFAT)家族成员的去磷酸化作用,使NFAT不能激活和刺激白细胞介素-2(IL-2)、IFN-γ等淋巴因子的基因表达,对T淋巴细胞起功能性阻断作用。但IL-2及NFAT-1基因敲除动物的改变与CsA的作用并不相符。IL-2基因敲除动物出现T淋巴细胞增生、骨髓成熟髓系细胞大量减少、B淋巴细胞分化发育受阻及自身免疫性疾病等表现。在体外,NFAT-1分子基因敲除动物细胞对李斯特杆菌、卵清蛋白的免疫反应增强;在体内,NFAT-1分子基因敲除动物发生过敏性炎症反应、血清IgE水平增高,说明NFAT-1可能对免疫系统起负性调节作用。研究提示CsA还可能通过刺激转化生长因子-β(TGF-β)产生而起作用,TGF-β也可抑制免疫,且CsA所致的不良反应与TGF-β类似,CsA对癌细胞生长的影响在加入TGF-β中和性抗体后可以逆转。
单药ALG/ATG治疗再障的有效率约45%,CsA对50%ALG/ATG耐药的患者有效。法国学者对94例重型AA(severeAA,SAA)患者组织了多中心前瞻性ALG和CsA对照治疗研究,3个月无效患者接受对照方案治疗。两组间3个月有效率为11.6%和16.0%,1年有效率(含接受对照方案治疗者)为31.6%和30.0%,1年生存率为70%和64%[9]。但该对照研究中两组患者接受对照治疗过早,不能完全确定为单药治疗的疗效。
由于ALG/ATG、CsA通过不同机制起作用,且交换使用能治疗各自耐药患者,所以临床开展了联合应用ALG/ATG、CsA对再障行强化免疫抑制治疗(intensiveIST,IIST)的研究。德国学者报道了IIST(ALG+CsA)与ALG多中心前瞻性随机对照研究,其中轻型再障24例,SAA60例。IIST治疗组的3个月、6个月有效率均高于ALG组(p<0.05):65%和39%,70%和46%;IIST组SAA患者6个月有效率达ALG组的2倍:65%vs31%(p<0.05)。随访41个月,IIST组、ALG组生存率分别为64%、58%,其中SAA患者两组生存率分别为80%和44%]。美国国立卫生研究院(NIH)以ATG+CsA对51例SAA患者进行IIST,3个月和1年的有效率分别为67%和78%,1年和2年实际生存率为86%和72%。我们序贯使用ALG/ATG与CsA治疗了36例SAA,6个月有效率为50.0%,1年和2年生存率分别75.0%和72.2%。
欧洲骨髓移植协作组多中心前瞻性对照研究了IIST(ALG+CsA)和CsA对轻型再障的疗效。IIST组和CsA组6个月有效率分别为74%和46%(p<0.05),但1年生存率无区别,分别为93%和91%。我们比较了以长效睾酮单用(15例)及合用CsA(30例)对慢性再障的疗效,合用组有效率为83.3%,高于单用长效睾酮的60.0%,且前者有效患者的BFU-E产率明显高于后者(p<0.05),接近正常。
对于ATG+CSA治疗无效或复发的患者,应用第二次ATG治疗有效率约为50%~60%。NIH最近回顾性研究显示马ATG治疗无效者应用兔ATG治疗后6个月有效率为33%,应用马ATG治疗后复发患者的有效率为68%。由于缺乏前瞻性随机临床试验比较兔和马ATG的疗效,第二疗程ATG的选择依赖于第一次应用是否有严重不良反应,医生的用药习惯及可应用的药物。
第三疗程的ATG治疗是否有益?英国回顾性研究显示,所有7例第一或第二次ATG治疗有效后复发的患者,第三次ATG治疗有效。但11例前两个疗程无效的患者,第三次应用ATG后,只有2例有效。多次应用ATG可能会有严重全身反应和过敏,因此应用应十分慎重。第三次应用ATG的通常为复发患者,这提示这种患者可能需要维持免疫抑制作用,控制对造血的免疫异常攻击。对于难治性患者,需要另外选择方案。
对于应用ATG是否有年龄高限问题,最近在低危MDS患者应用ATG治疗的研究显示老年人应用ATG是安全的。60岁以上AA患者应用ATG与年轻患者同样有效,但感染和出血几率增加,使生存率下降。最近应用低剂量ATG的I/II期研究正在评价其在60岁以上患者的有效性和耐受性。既往体外实验显示应用标准剂量的淋巴细胞球蛋白的血清药物水平对骨髓造血祖细胞具有毒性。在12例患者中,只有1例部分有效,显示低剂量的ATG不具有标准剂量ATG的疗效。从实际应用角度上,老年人是否应用ATG应该根据患者的体能状态。60岁以上患者应用ATG前应进行详细的评估,尤其是高于70岁的患者。80岁以上患者通常不考虑应用ATG.
IST治疗无效的可能原因目前正在研究,被称为非免疫发病的AA、误诊或免疫攻击已经导致干细胞耗竭。另外,如果在仍然有免疫攻击的情况下,免疫抑制作用不强也可能是治疗无效的原因。ATG+CSA之外加用其他免疫抑制药物,或应用较ATG免疫抑制作用更强的药物如,大剂量CY和alemtuzumab。选用ATG和CSA不同作用机制的药物如骁悉,抑制次黄(嘌呤核)苷酸脱氢酶,这种酶是嘌呤合成所必需,尤其是在激活的淋巴细胞中。最近104例患者的研究应用ATG+CSA和骁悉联合治疗,6个月有效率为62%,复发率为37%,这与ATG+CSA治疗疗效相似。大多数复发患者(63%)在应用骁悉过程中复发。没有干细胞支持的大剂量CY能使AA完全缓解,但由于血象恢复缓慢,侵袭性真菌感染率高影响了其广泛应用。17例ATG或CSA治疗无效的患者,53%脱离输血,59%患者生存长于29个月。然而,CY免疫抑制增强的同时并没有阻止2例患者发生晚期克隆性疾病,在所有伴PNH克隆的患者均没有根除异常克隆。Alemtuzumab免疫抑制作用强度和时间较ATG强,目前正在初治和难治患者中进行试验。难治性AA的治疗十分棘手,目前缺乏强有力预测试验以预测IST治疗的有效率,缺乏新的免疫抑制药物。重组人抗IL-2受体单抗(daclizumab)在未治疗非重型AA具有一定效应,有效与CD8+T细胞呈正相关。
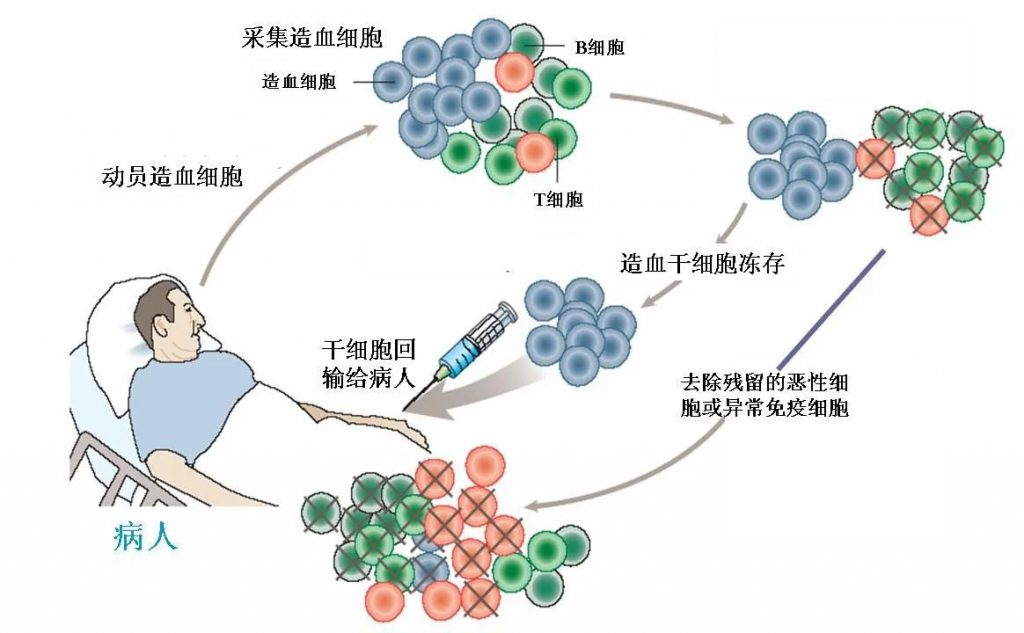
2、HLA相合同胞供者BMT
AA患者BMT基本上都不是前瞻性的,大多数资料来源于欧洲或国际骨髓移植登记处(EBMT或IBMTR)或单中心的资料。对于年轻重型再障,早期确定患者是否有相合的同胞供者至关重要。如果有合适的同胞供者,患者应不选择IST,除非有医学或个人原因不能进行移植。目前应用的最广泛的预处理方案为环磷酰胺(CY)200mg/kg和ATG+CSA,甲氨蝶呤(MTX)预防GVHD。预处理方案中加入ATG的优势还需要进一步证实。HLA相合同胞供者BMT的长期总生存率为80%~90%。不同年龄的生存率存在差异,年青患者较好。但是移植成功后,慢性GVHD仍然是棘手的问题。移植排斥率约为5~15%。重度输血患者排斥率高,这提示AA患者应在多次输血使患者对HLA或非HLA抗原致敏前行移植至关重要。G-CSF动员外周血干细胞已经被用于增加干细胞的数量、加速中性粒细胞和血小板的恢复及减轻移植物排斥方面。然而回顾性分析国际血液和骨髓移植研究中心(CIBMTR)/EBMT结果显示,AA应用外周血干细胞移植,移植排斥没有减少,慢性GVHD更多,预后差。这样,目前仍然认为骨髓是干细胞移植的最佳来源。氟达拉滨联合减量CY+ATG目前正用于克服移植物排斥,尤其是在致敏患者。NIH应用此方案治疗重度致敏患者,取得了良好的植入率,但急性和慢性GVHD均较高。氟达拉滨为基础的方案需要进行大规模多中心研究验证,它可能更适合老年人。在移植的AA的患者,需要考虑生育问题,大剂量CY对生育影响不大,但应该避免放射。氟达拉滨为基础预处理方案对于生育的影响还要进一步研究。CSA的应用降低了急性GVHD的发生率和严重性,避免了放射。但慢性GVHD仍然发生于25~40%的患者,是导致死亡的重要原因。最近SaintLouisHospital研究资料CY/ATG联合全腹照射方案,慢性GVHD发生率为40%,其中80%为广泛型。慢性GVHD的危险因素包括急性GVHD、放射和年龄大。Seattle研究发现骨髓细胞超过3.4×108有核细胞/kg也是慢性GVHD的危险因素,但没有关于CD34+细胞的研究。抗CD52单克隆抗体(Campath)的应用使急性GVHD(14%)和慢性GVHD(4%)发生率明显下降。虽然总移植排斥率为24%,骨髓输注前应用Campath较输注前后分别应用Campath,排斥率下降16%。死亡率随年龄增长而升高。40岁以上患者生存率只有50%。因此,只有40岁以下患者才考虑将BMT作为一线治疗方案。BMT也要根据患者体能状态及其意愿。预处理方案应用氟达拉滨+低剂量CY(1200mg/m2)+ATG适合于年龄较大的患者,EBMT目前也将该方案应用于相合无关供者(MUD)BMT。其他预后不良的因素包括以前应用过ATG或CSA或雄激素、诊断到移植超过1年和应用放疗。其他预后好的因素包括血制品供应提升和质量的提高、更好的抗生素、抗真菌及抗病毒药物的使用。虽然不能被统计学所证实,但生存率的提高是最主要的。应该避免放疗,因为其有不育和第二肿瘤的危险性。对于伴有TERC或TERT突变的成人获得性AA(亚临床DC),BMT是唯一治愈疾病的方法。减量的预处理方案能降低这类患者BMT后的晚期并发症。
3、相合无关供者BMT
CIBMTR回顾性分析1988~1998间重型AA移植的结果显示,MUDBMT疗效差(5年生存率39%),移植排斥率高,GVHD高,感染发生率高。随着时间的进展,疗效无明显提高。这个研究中,HLA配型依赖于低分辨DNA配型HLA-A、-B和-DR位点。一项日本研究应用高分辨DNA配型技术发现HLA-A、-B和-DRB1MUD生存率提高(60%)。
最近研究试图应用低剂量全身照射或无照射的氟达拉滨为基础方案,以降低移植排斥,提高生存率。在多中心前瞻性研究中,Deeg等报道应用减低剂量的全身照射方案,87例无关供者总移植排斥率只有5%(相合者2%,不相合者11%)。但肺毒性和GVHD仍然是个问题。EBMT应用氟达拉滨+低剂量CY(1200mg/m2)+ATG结果显示移植排斥率高(18%),但急性和慢性GVHD发生率低。EBMT也应用alemtuzumab替代ATG的方案,或G-CSF动员的CD34+选择性PBSC。然而,其疗效还有效证实。
因为脐血移植的优点是对HLA配型要求不严,所以在没有完全相合骨髓供者时,可以考虑脐血移植。目前发表的脐血移植治疗获得性AA的资料很有限。脐血中细胞数量很少,这使其在AA中应用成为问题,因为AA需要足够的良好干细胞以获得移植的成功。最近应用双份UCB移植提高干细胞数量,治疗高危MDS/AML,取得了较高的植入率。对于个体而言,两份中只有一份能长期植入。植入单位中发现CD3+细胞数量升高,而不是有核细胞或CD34+细胞。
4、再障患者的生存质量(QOL)
随着BMT和IST治疗AA水平的提高,两者在总生存时间上的差异已经没有明显差异,因此,就体现出无失败生存(FFS)和生存质量(QOL)的重要性。一项研究对1976~1999年54例移植患者和154例IST患者回顾性研究表明,总生存(OS)和无事件生存(EFS)相似。但IST有以下缺点①由于药物毒性导致的症状时间长;②依赖输血的时间长;③部分缓解的时间长;④继发克隆性改变的时间长。移植者完全缓解的时间长,无症状的时间长,但伴有广泛慢性GVHD的缺点。
5、妊娠期AA的处理

少数情况下,AA可以发生于妊娠期间,并且在分娩后或终止妊娠后自发缓解。但并不是所有妊娠期患者都这样。没有证据显示获得性AA应用ATG和CSA治疗后生育能力下降,但三分之一以上的妊娠期再障患者会复发,无论其是否缓解。是否继续妊娠由患者自己决定。应用大量输血支持治疗原发或继发性妊娠期AA,可能增加红细胞、HLA和血小板抗原的自身免疫。肾移植研究显示CSA对于婴儿没有影响,因此妊娠期间可继续应用CSA治疗。ATG很少用于妊娠期AA的治疗。与IST治疗患者的经验相比,成功移植后妊娠并不导致AA的复发。
文章属于病友个人治疗见解及自身对疾病的治疗分析仅供参考(不代表本站同意其说法)谨慎参阅!
提示:ITP6病友网站内任何对疾病的建议都不能替代执业医师当面诊断!
作者:血小板减少交流群病友分享(文章内图片于群内交流)
链接:https://www.itp6.com/7338.html
文章版权归作者所有,未经允许请勿转载。
 微信扫一扫
微信扫一扫 