爸爸发现12岁的小柯(化名)肚子明显比其他同龄孩子的大一些,而且时间很长了都不见“消退下去”,于是开始带着孩子四处求医。来到我院后,分诊台考虑小柯“脾肿大”分检去血液肿瘤科就诊,血液肿瘤科王华主任医师为孩子进行了详细的体检,并细致的询问了孩子的病史,经验丰富的王华主任医师立刻建议孩子进行骨髓穿刺。
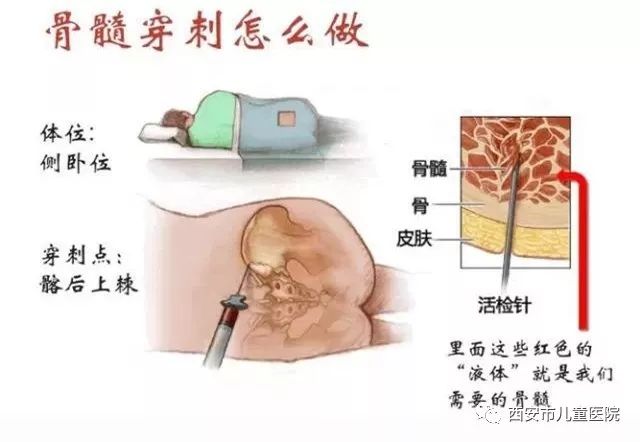

很多人问小编,骨髓穿刺用的针那么粗,会不会特别疼?其实并没有那么恐怖,疼痛感和一般注射差不多,甚至不如打麻药疼,抽取时间在1分钟左右,之后立刻就可以下床行走了,按压10分钟左右,不过因为伤口不能沾到水,3天内都不宜洗澡,麻药2-3小时后消失,只会略有酸疼感。
小柯的血常规如下:白细胞(6.85*10^9/L),红细胞(4.31*10^12/L),血红蛋白119g/L,血小板126*10^9/L。镜下白细胞百分比:淋巴细胞30%,单核细胞5%,中性粒细胞65%,粒细胞可见中毒颗粒,提示存在感染。
小柯的血涂片,其中较为粗大 、分布不均的深蓝色或蓝黑色颗粒(因为拍摄器材显像不同,有时候也偏紫色)就是中毒颗粒
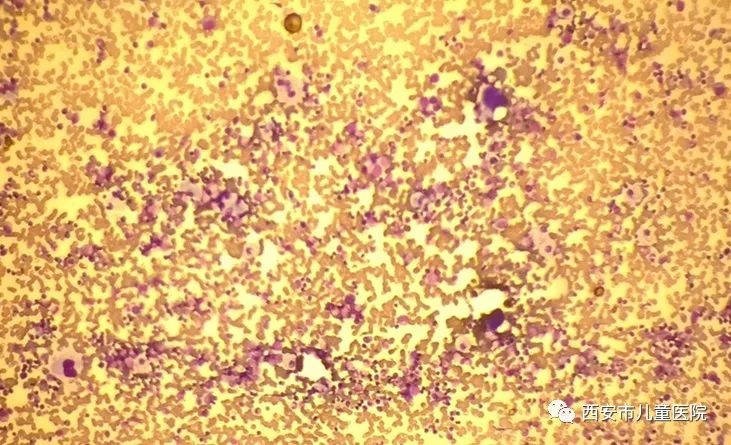
我院中心实验室在骨髓低倍镜和骨髓油镜发现了戈谢细胞。
什么是戈谢细胞呢?这种细胞胞体大,直径达20~100μm,形态为卵圆形或多边不规则形;胞核较小偏位,圆或椭圆形,1~3个或更多,染色质粗糙,副染色质明显,偶见核仁;胞质丰富,淡兰色,无空泡,胞质中含有许多与细胞长轴平行的粗暗条纹样结构,交织成网,如洋葱皮样或蜘蛛网状。骨髓中见到戈谢细胞就可以确诊小柯就是戈谢病了!
戈谢病是种什么病?
戈谢病(Daucher disease,旧称高雪病)是一种“罕见遗传病”,为常染色体隐性遗传。
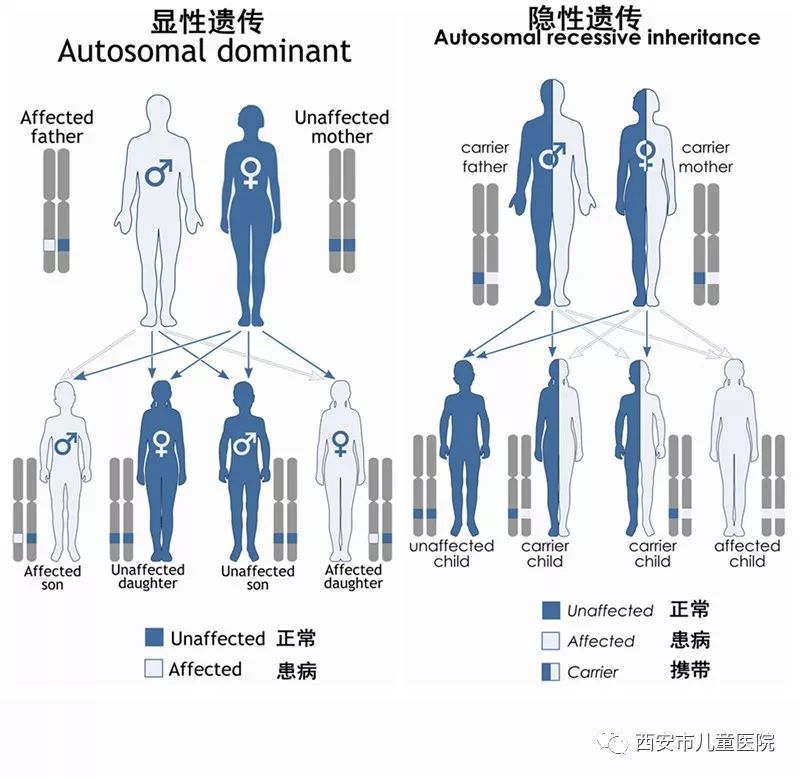
由于基因突变致机体β葡萄糖脑苷脂酶活性缺乏,这种酶帮助人体分解名为葡糖脑苷脂的脂肪。但有患有戈谢病的患儿,不能产生这种β葡萄糖脑苷脂酶,因而也不能分解脂肪,这样脂肪就会逐渐累积在肝脏、脾和骨髓中,体现在孩子身上就是“肝、脾肿大”直至肚子肿大胀起。
戈谢病发病率极低,为罕见病,但在临床极易误诊、漏诊,目前中国有记录的患儿不到1000人。戈谢病能导致疼痛、疲劳、黄疸、骨损伤、贫血甚至死亡。一般情况下,这种病先天性的代谢异常,虽然各个年龄均可发病,大部分患者很小的时候(7岁以下)就会发病,而小柯12岁才来就诊,发现算是相当晚了,如若没有做骨髓穿刺,他可能还辗转于其它各种检查未必可以确诊,我院中心实验室的骨髓细胞形态学检查在该类疾病诊断中发挥了极大的作用。
戈谢病容易误诊还因为,有时在骨髓检查中可能会看到一种与戈谢细胞非常相似的假戈谢细胞(pseudo-gaucher’s cell),它可出现在慢性粒细胞白血病(CML)、地中海贫血、多发性骨髓瘤、霍奇金淋巴瘤、浆细胞样淋巴瘤(plasmocytoid lymphoma)及慢性髓性白血病。它与戈谢细胞的不同点是胞质中无典型的管样结构,鉴别诊断时可做GC酶活性测定。
戈谢病根据各器官受累的程度、发病的急缓,以及有无神经系统受累,分为3型:
Ⅰ型(成人型)——慢性型又称非神经型。临床上相对最常见,尤其是犹太人种发病率高(德裔犹太人Ⅰ型戈谢病发病率接近千分之一,远高于其他人种低于二十万分之一的发病率),儿童与成人均可发病,以学龄前儿童发病者多,起病缓慢,病程长,无神经系统受累症状。发病越早,酶活力越低。通常Ⅰ型患儿的β葡萄糖脑苷脂酶活力相当于正常人的12%~45%。Ⅰ型戈谢病进展缓慢,脾切除后可长期存活,智力正常,只是生长发育落后于健康人,酶替代治疗效果显著,预后最好。
Ⅱ型(婴儿型)——急性型又称神经型。较多在1岁以内发病,最早于生后1~4周出现症状,病情随起病早晚而不同,除Ⅰ型的症状、体征外,神经系统症状明显,此型的β葡萄糖脑苷脂酶活力最低,几乎难以测出。Ⅱ型戈谢病多于发病后1年内死于继发感染,只有少数可以存活2年以上。
Ⅲ型(幼年型)——亚急性型也称神经型。起病较Ⅱ型缓慢,可在婴幼儿期发病,除内脏受累外,可有1项轻、中度神经系统表现。多数在10岁左右出现,此型智力障碍相对较轻。
提示:遗传性代谢性疾病产前诊断是防止遗传病发生的有效措施,本症确定患儿基因型后,其母再次妊娠可做产前基因诊断,也可予杂合子检查。
随着分子生物学及生物制药等领域的进展,对戈谢病的诊断及治疗已经有了长足进步,目前已成为遗传代谢病中为数不多的可治性疾病之一。近年来,国内通过脐带血配型,已经成功为数例较为严重的戈谢病患儿移植脐带血造血干细胞进行治疗。
除了治疗血液系统疾病外,脐带血还可治疗免疫系统、遗传代谢性及先天性疾病等。在我国脐带血造血干细胞治疗技术已应用于白血病、骨髓增生异常综合征、恶性淋巴瘤、多发性骨髓瘤、恶性肿瘤、重症再生障碍性贫血、重症放射病、骨髓衰竭、血红蛋白病、重症免疫缺陷病、代谢性疾病等10余类。
文章属于病友个人治疗见解及自身对疾病的治疗分析仅供参考(不代表本站同意其说法)谨慎参阅!
提示:ITP6病友网站内任何对疾病的建议都不能替代执业医师当面诊断!
作者:血小板减少交流群病友分享(文章内图片于群内交流)
链接:https://www.itp6.com/12754.html
文章版权归作者所有,未经允许请勿转载。
 微信扫一扫
微信扫一扫 